联系我们 +0512-62607321 or rpxds@rpxds.com
筛选-优化-验证
开放 AI 设计 联合开发模式
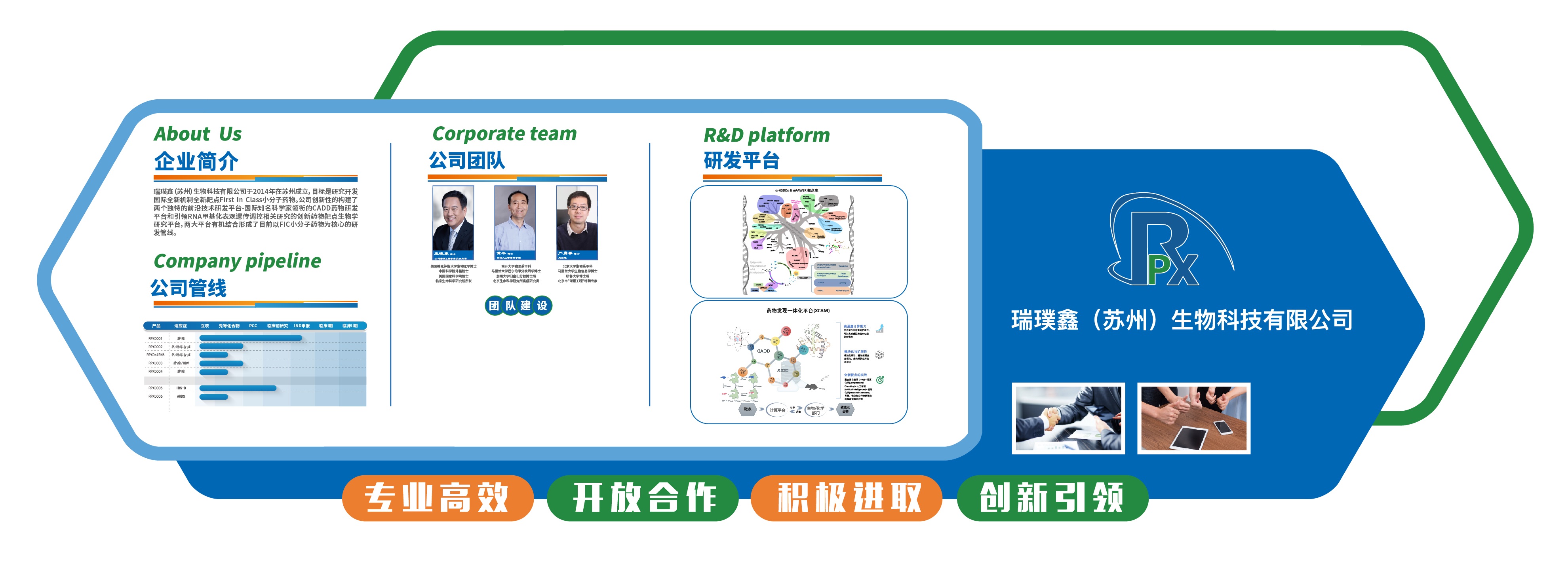
2014初创研发型药企,以结构计算驱动,转化肥胖癌症等原创新药
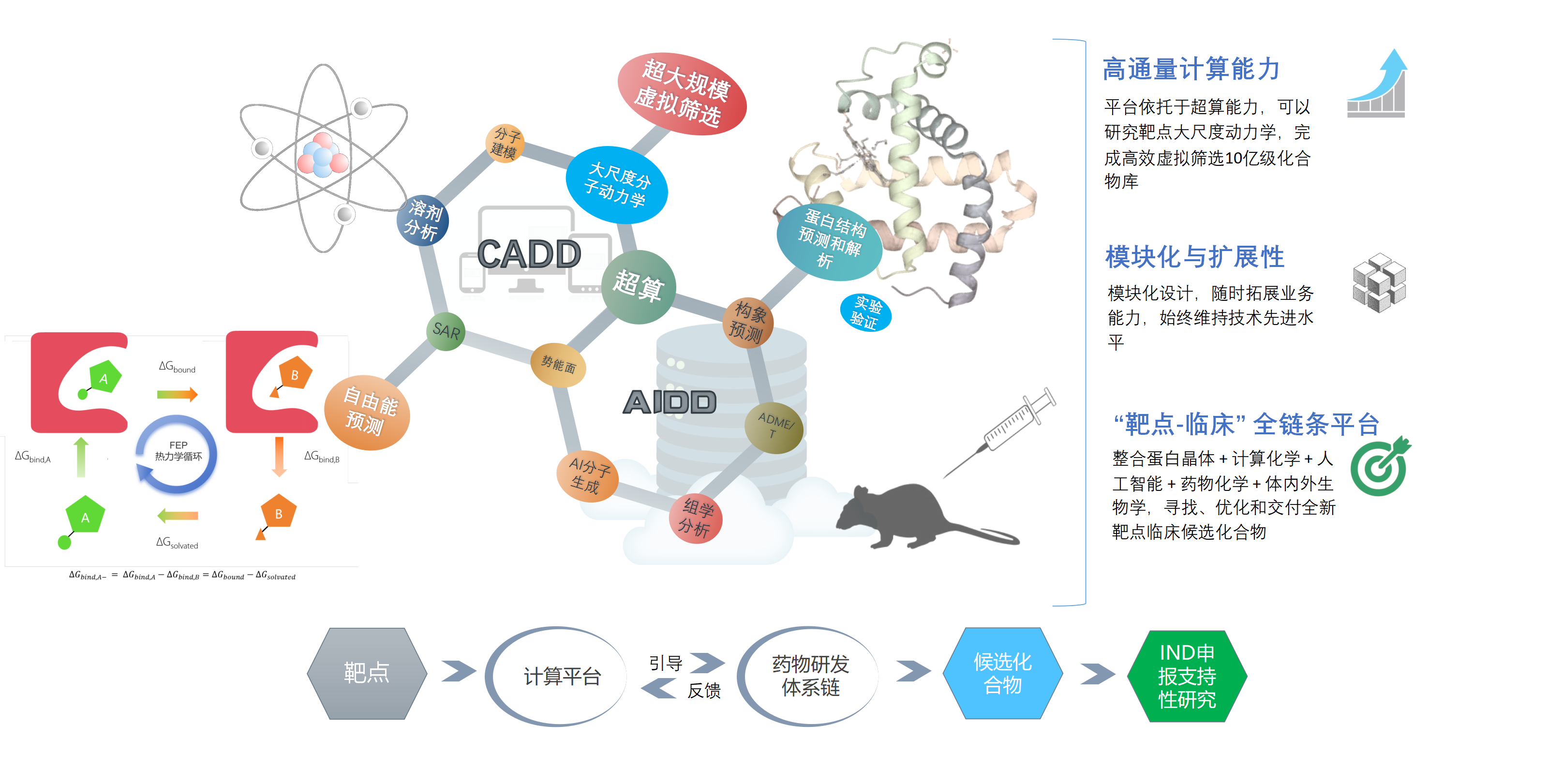
结构生物学+计算化学+生化结构细胞动物一体化设计
基础发现直转临床前,布局靶向FTO药物
创始人黄牛博士领军,结构计算与转化医学复合团队加速新药孵化
Copyright (c) 2014 - 2026, 瑞璞鑫版权所有. 苏ICP备17018049号
Bootstrapious. DevCows.